If you were giving a lot more midazolam (Versed) by intramuscular injection to stop a seizure and the seizure just would not stop, or got worse, maybe you were giving ondansetron (Zofran).
If you were giving a lot more midazolam by injection to sedate a patient and the sedation just wasn’t having its usual effect, maybe you were giving ondansetron. While rare, there can be very serious side effects from too much ondansetron.
Dose-dependent serious cardiac arrhythmias may be observed with higher dosages of ondansetron in those patients with certain pre-existing cardiac conditions. Patients may also be at risk for serotonin syndrome. Serotonin syndrome is associated with increased serotonergic activity in the central nervous system. Most reports of serotonin syndrome have been associated with concomitant use of certain drugs, some commonly used during surgery, such as fentanyl. Some of the reported cases of serotonin syndrome were fatal.[1]
How do you recognize serotonin syndrome?
Serotonin syndrome (SS) is a group of symptoms that may occur following use of certain serotonergic medications or drugs. [1] The degree of symptoms can range from mild to severe.[2] Symptoms include high body temperature, agitation, increased reflexes, tremor, sweating, dilated pupils, and diarrhea.[1][2] Body temperature can increase to greater than 41.1 °C (106.0 °F).[2] Complications may include seizures and extensive muscle breakdown.[2] [2]
2 mg of midazolam is much too low a dose to try to stop a seizure, unless it is the only packaging you have and you are giving 5 intramuscular injections at a time. The best response to prehospital treatment of seizures was by giving 10 mg of intramuscular midazolam to adults (over 40 kg) and 5 mg of intramuscular midazolam to children (under 40 kg).
Maybe you think that is too much midazolam. The highest quality and largest pre-hospital study does not support using lower doses.
Our data are consistent with the finding that endotracheal intubation is more commonly a sequela of continued seizures than it is an adverse effect of sedation from benzodiazepines.11 [3]
There are other uses for midazolam, so you should be aware of the possibility that what you think is midazolam is really ondansetron.
Are the syringes labeled incorrectly for the contents?
Fresenius Kabi USA is voluntarily recalling Lot 6400048 of Midazolam Injection, USP, 2 mg/2 mL packaged in a 2 mL prefilled single-use glass syringe to the hospital/user level. The product mislabeled as Midazolam Injection,
USP, 2 mg/2 mL contains syringes containing and labeled as Ondansetron Injection, USP, 4 mg/2 mL.[1]
Based on that, the syringes should be correctly labeled as ondansetron, but they are in blister packs labeled as containing midazolam or they are in boxes of blister packs listed as containing midazolam or both or something else.
If you use this packaging of midazolam, check the lot number, the syringe, and any other labels to make sure that they all agree.
What if you need some ondansetron pre-filled syringes?
Send them back anyway. Maybe only some of the syringes are labeled correctly.
What do the syringes look like?
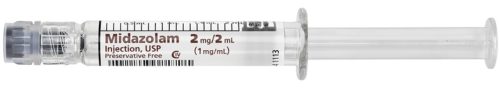
What does the ondansetron syringe look like? This one is with a blister pack.

There are other possibilities for mislabeling that could be much more harmful, so read the syringe before you push anything by any manufacturer.

That probably would not be as harmful as it seems, because it would be pushed slowly, so it might be metabolized as quickly as it is pushed. The ones below would still be expected to produce a much greater respiratory depression than even an extreme midazolam respiratory depression.
–
Footnotes:
–
[1] Fresenius Kabi Issues Voluntary Nationwide Recall of Midazolam Injection, USP, 2 mg/2 mL Due to Reports of Blister Packages Containing Syringes of Ondansetron Injection, USP, 4 mg/2 mL
For Immediate Release
November 3, 2017
Voluntary Recall
Recall announcement
–
[2] Serotonin syndrome
Wikipedia
Article
–
[3] Intramuscular versus intravenous therapy for prehospital status epilepticus.
Silbergleit R, Durkalski V, Lowenstein D, Conwit R, Pancioli A, Palesch Y, Barsan W; NETT Investigators.
N Engl J Med. 2012 Feb 16;366(7):591-600.
PMID: 22335736 [PubMed – in process]
Free Full Text from N Engl J Med.
.












Subscribe to RogueMedic.com